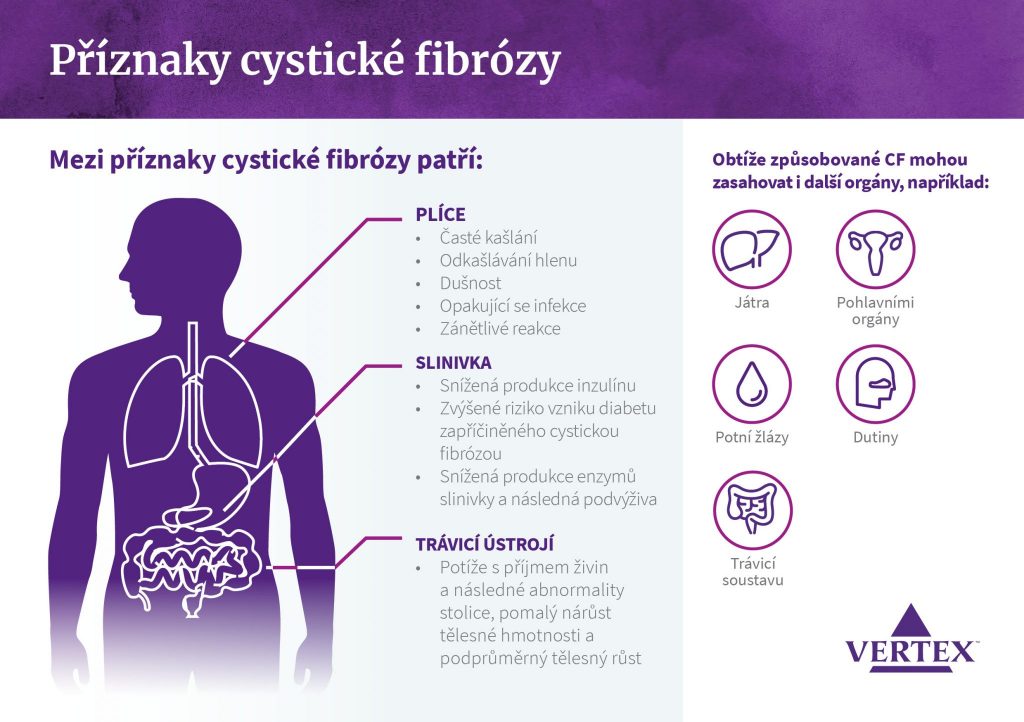
Cystická fibróza (CF) je jedním z nejrozšířenějších vzácných onemocnění v České republice, kde jí trpí necelých 700 pacientů[i]. Po celém světě pak žije přibližně 75 tisíc osob s touto nemocí.[ii] Jedná se o postupně se zhoršující geneticky podmíněné onemocnění, které způsobuje tvorbu lepivého hlenu v plicích a trávicím ústrojí.[iii]
Rozvoj cystické fibrózy způsobuje chybný nebo chybějící protein CFTR v důsledku mutace genu zodpovědného za jeho produkci. Výsledkem jsou problémy při přechodu chloridových iontů (součást kuchyňské soli) přes membránu buněk v těle. To vede k tvorbě a hromadění hustého a lepivého hlenu v plicích a dalších orgánech.
CF Foundation. 2020. About Cystic Fibrosis. K dispozici online na adrese: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/ [Použito v dubnu 2020].
CF Foundation. 2020. Cystic Fibrosis-Related Diabetes. K dispozici online na adrese: https://wwwcff.org/Life-With-CF/Daily-Life/Cystic-Fibrosis-Related-Diabetes/ [Accessed April 2020].
Rozvoj onemocnění cystickou fibrózou může ovlivňovat život pacientů v mnoha ohledech
- Delší pobyty v nemocnici
- Produktivita
- Duševní zdraví
- Rodinný život
- Společenské vztahy a uplatnění

Cure4CF. 2020. Living with Cystic Fibrosis – Cure4CF. K dispozici online na adrese: https://cure4cf.org/living-with-cystic-fibrosis/ [Použito v dubnu 2020].
Marshall, B., et al. 2019. Cystic Fibrosis Foundation Patient Registry 2018 Annual Data Report. Bethesda, Maryland: Cystic Fibrosis Foundation. K dispozici online na adrese: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf [Použito v dubnu 2020].
Yusen, R.D., et al. 2015. The Registry of the International Society for Heart and Lung Transplantation: thirty-second official adult lung and heart-lung transplantation report—2015; focus theme: early graft failure. The Journal of Heart and Lung Transplantation, 34(10), str. 1264-1277.
Dopady mutace genu CFTR
Normální protein CFTR
Tyto proteiny tvoří za normálních okolností součást membrány buněk na povrchu plic, orgánů trávicí soustavy, potních žláz a dalších orgánů. Jejich úkolem je regulovat přenos chloridových iontů a vody do buněk a z buněk ven.
Poškozený nebo chybějící protein CFTR
V případě mutace genu CFTR nejsou proteiny CFTR vytvořené na základě informace z takového genu schopné zaujmout správné místo v buněčné membráně nebo správně fungovat. Tím dochází ke zhoršení přenosu iontů a vody do buňky a z buňky a ke vzniku hustého, lepivého hlenu, který následně omezuje průchodnost dýchacích cest a dalších orgánů.
Důsledky
Nedostatečný přenos chloridových iontů a vody přes buněčnou membránu způsobuje dehydrataci povrchu postižených buněk. Hlen, který je normálně řídký a volně se po povrchu buněk pohybuje, houstne a omezuje průchodnost orgánů a dýchacích cest. Navíc tím vzniká vhodné prostředí pro množení bakterií, což vede k infekcím a zánětům.
Mohlo by vás zajímat


CF Foundation. 2020. Types of CFTR Mutations. K dispozici online na adrese: https://www.cff.org/What-is-CF/Genetics/Types-of-CFTR-Mutations/ [Použito v dubnu 2020].
Reference, G., 2020. CFTR Gene. [online] Genetics Home Reference. K dispozici online na adrese: http://ghr.nlm.nih.gov/gene/CFTR [Použito v dubnu 2020].
Ensinck, M., et al. 2020. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells, 9(3), str. 754.
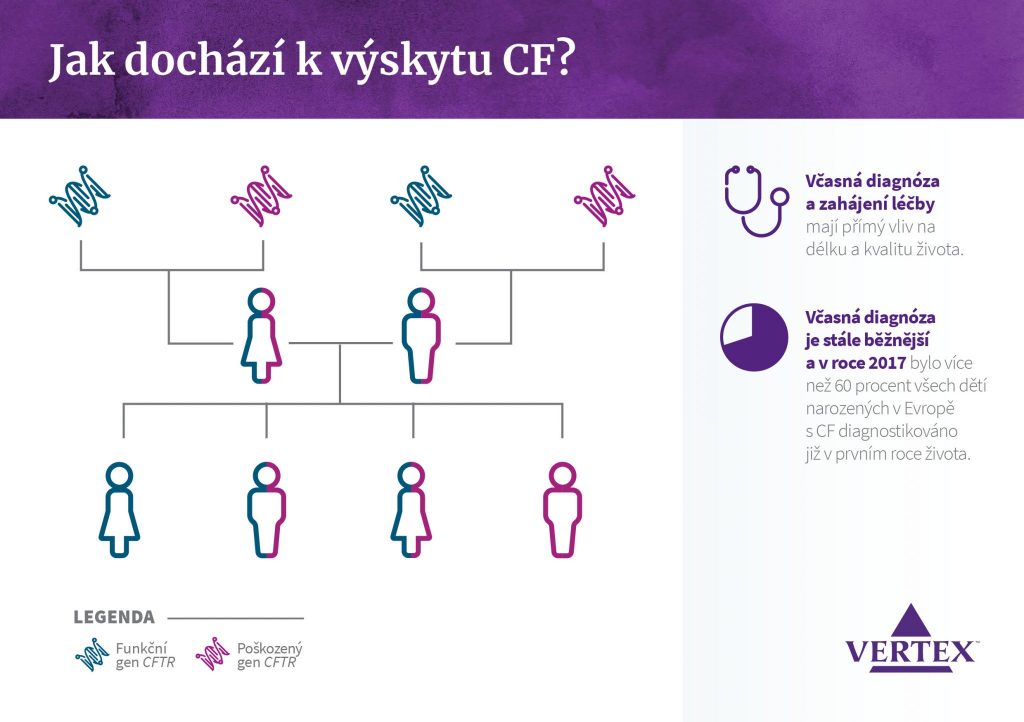
Jak dochází k výskytu CF?
Aby člověk trpěl cystickou fibrózou, musí mít dvě poškozené kopie genu CFTR (CF je autozomálně recesivní porucha). Přenašečem CF je osoba s jedním poškozeným genem CFTR získaným od jednoho z rodičů a jedním funkčním genem od druhého.
Aby došlo k rozvoji CF, je nutné zdědit dva poškozené geny CFTR – od každého rodiče jeden.
Včasná diagnóza a zahájení léčby mají přímý vliv na délku a kvalitu života.
Včasná diagnóza je stále běžnější a v roce 2017 bylo více než 60 procent všech dětí narozených v Evropě s CF diagnostikováno již v prvním roce života.
CF Foundation. 2020. About Cystic Fibrosis. K dispozici online na adrese: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/ [Použito v dubnu 2020].
Zolin, A., et al. 2019. ECFS Patient Registry Annual Data Report 2017. K dispozici online na adrese: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports [Použito v dubnu 2020].
Tridello, G., et al. 2018. Early diagnosis from newborn screening maximises survival in severe cystic fibrosis. ERJ Open Research, 4(2).
CZ-02-2100001
[i] Český registr cystické fibrózy, 2020. K dispozici na adrese: https://cfregistr.cz/statistika/
[ii] Jabar, A., et al. 2014. New and Evolving Therapies for Cystic Fibrosis Patients. Pediatric Allergy, Immunology, and Pulmonology, 27(2), str. 92-94.
[iii] CF Foundation. 2020. About Cystic Fibrosis. [online] K dispozici na adrese: <https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/>
RESERVED, I., 2020. Orphanet: Cystic Fibrosis. [online] K dispozici na adrese: <https:// www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=586.>.
Článek vychází v partnerství se společností Vertex.